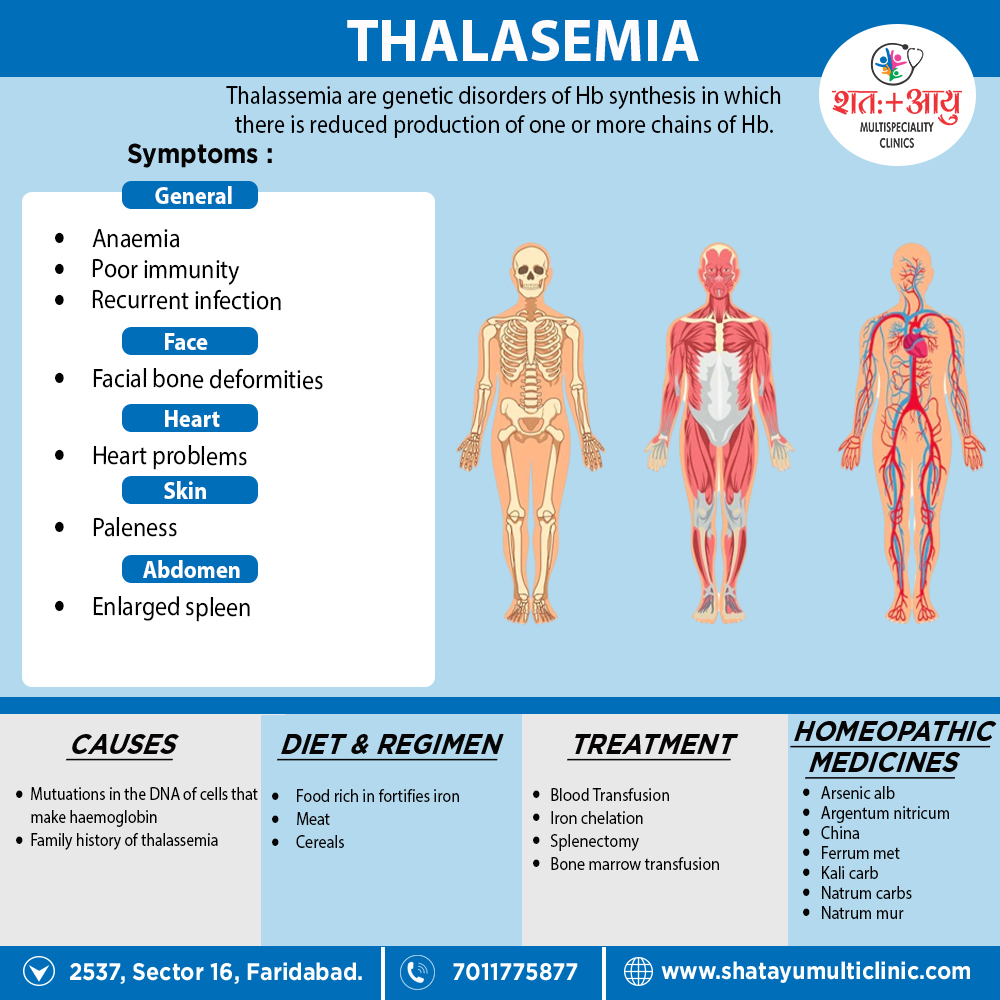
Homeopathic Treatment of Thalassemia
Homeopathy treats the person as a whole. It means that homeopathic treatment focuses on the patient as a person, as well as his pathological condition. The homeopathic medicines selected after a full individualizing examination and case-analysis.
which includes
- The medical history of the patient,
- Physical and mental constitution,
- Family history,
- Presenting symptoms,
- Underlying pathology,
- Possible causative factors etc.
A miasmatic tendency (predisposition/susceptibility) also often taken into account for the treatment of chronic conditions.
What Homoeopathic doctors do?
A homeopathy doctor tries to treat more than just the presenting symptoms. The focus is usually on what caused the disease condition? Why ‘this patient’ is sick ‘this way’?.
The disease diagnosis is important but in homeopathy, the cause of disease not just probed to the level of bacteria and viruses. Other factors like mental, emotional and physical stress that could predispose a person to illness also looked for. No a days, even modern medicine also considers a large number of diseases as psychosomatic. The correct homeopathy remedy tries to correct this disease predisposition.
The focus is not on curing the disease but to cure the person who is sick, to restore the health. If a disease pathology not very advanced, homeopathy remedies do give a hope for cure but even in incurable cases, the quality of life can greatly improved with homeopathic medicines.
Homeopathic Medicines for Thalassemia:
The homeopathic remedies (medicines) given below indicate the therapeutic affinity but this is not a complete and definite guide to the homeopathy treatment of this condition. The symptoms listed against each homeopathic remedy may not be directly related to this disease because in homeopathy general symptoms and constitutional indications also taken into account for selecting a remedy.
Medicines:
Alumina
- Generally, Anaemia and chlorosis in young girls at puberty.
- Menses pale also scanty, with abnormal cravings for indigestible things.
- Lastly, Profuse aluminous leucorrhoea.
Argentum nitricum
- Shortness of breath, without either lungs or heart being affected.
- Furthermore, Sallow complexion from defective oxidation of the blood.
- Heart- burn, dyspepsia; irritative flatulent gastralgia; additionally, round ulcer of stomach.
- Menses irregular, either scanty or copious; spinal irritation, albuminuria, tendency to diarrhoea.
- Constant desire especially for candy or sugar.
China
- Complaints specifically from loss of animal fluids, be it blood, semen, diarrhoea, leucorrhoea or over lactation.
- Moreover, Great debility, trembling, aversion to exercise.
- Palpitations with rush of blood to head, additionally redness of face with cold hands.
- Heaviness of head, with loss of sight, fainting also ringing in ears.
- Sleeplessness; In detail, intolerance of fruits.
Ferrum met
- Basically, Pure anemia with appearance of false plethora.
- Face ashy either pale or greenish, becomes bright red in flushes, great paleness of mucous membranes.
- Bellow’s sound of the heart also anaemic murmurs of the arteries and veins.
- Vomiting as soon as food is taken, with relief of gastralgia pains, also prostration with lethargic dullness.
- Animal food not desired, nor is it well borne by the stomach if taken into it.
- Lastly, Anaemia of chlorotic girls and women.
Kali carb
- Frequent chilliness, every time patients goes out of doors.
- Patient becomes chilly from deficiency of red blood-corpuscles in the blood.
- Vertigo when turning head rapidly or from riding in a carriage, with humming in ears.
- Weakness of sight from sexual indulgence.
Natrum Mur
- Blood impoverished; anaemia from loss of fluids; additionally malarious cachexia; emaciation.
- Skin harsh, dry, yellow; great exhaustion from any little exertion of either mind or body.
- Palpitation, with sensation as if a bird’s wing were fluttering in left chest.
- Pressure and distension of stomach; constipation, with contraction of anus; terrible sadness.
Natrum carb
- Pallid anaemia, with great debility, Milky-white skin.
- Vitality below par; emaciation; nervousness also anxiety.
- Aggravation during a thunder storm.
- Playing on either piano or hearing music makes her nervous.
- Inertia in psoric, phlegmatic persons.